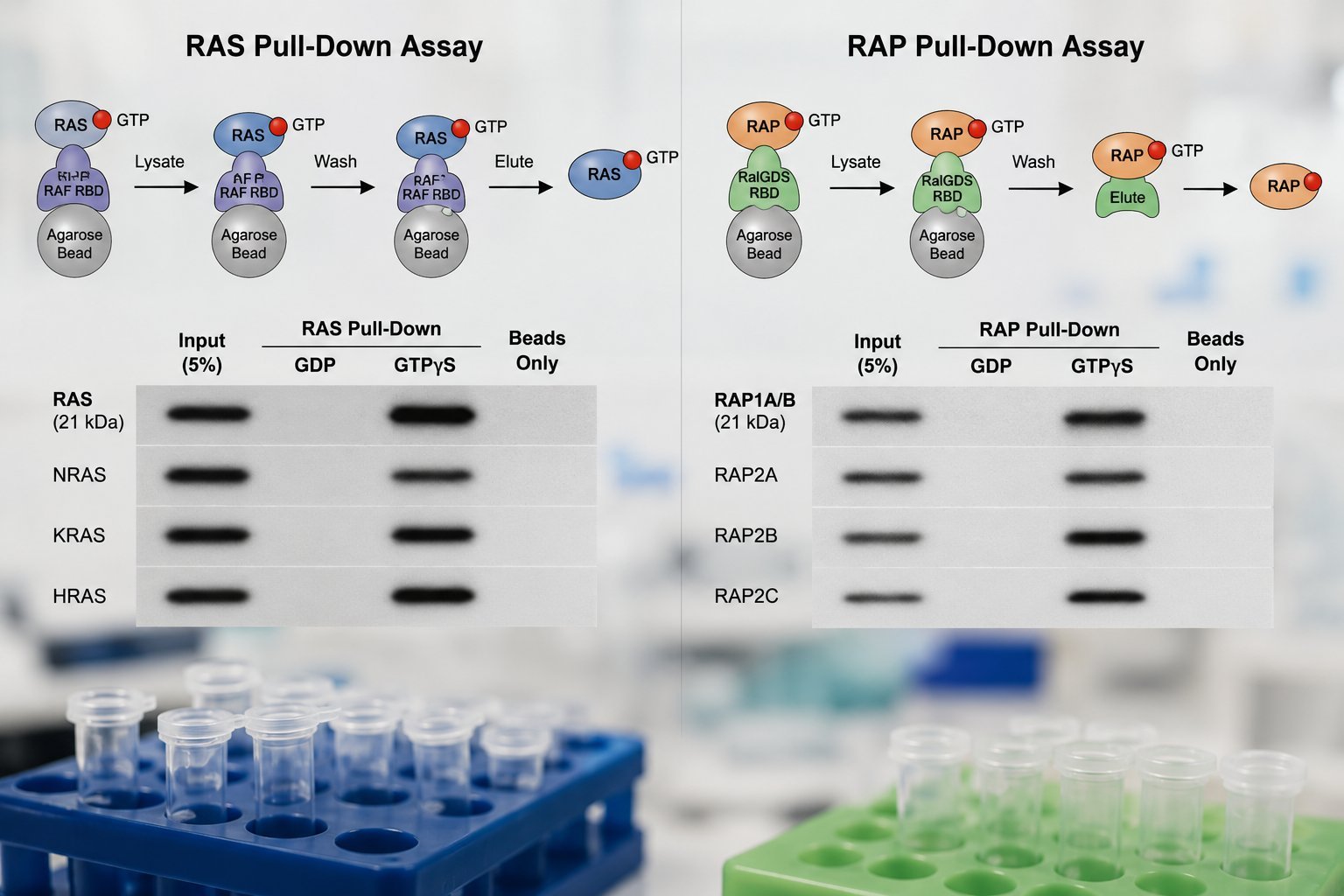
Ras/Rap activation assays
GST pull-down assays to detect active Ras or Rap in transfected cells, using a GST fusion of the Ras/Raf binding domain (RBD) or RalGDS RBD. Includes buffer recipes and an alternate Ras binding assay after Kelley et al. (EMBO J. 2001).
Ras/Rap pull-down assay
A) Preparation and purification of GST-Ras/Raf binding domain or RalGDS GST-RBD
- Inoculate 1 litre of LB broth with 15 ml of an overnight culture of E. coli containing the GST-RBD construct. Incubate for 2½ hours at 37 °C with shaking, then induce protein expression with 1 mM IPTG for 3 hrs at 37 °C.
- Pellet cells at 5000 rpm (Beckman JA-14 rotor) for 15 minutes and discard supernatant.
- Resuspend pellet in ~50 ml of ice cold PBS, 1% (v/v) Triton X-100, 1 mM EDTA and sonicate cells on ice (3× 30 sec bursts with 45 sec cooling periods). Incubate on ice for 15–30 min.
- Centrifuge lysate at 18000 rpm (Beckman JA-20 rotor) for 30 min. Freeze immediately on dry ice in 500 µl aliquots and store at −80 °C.
- For each pull-down sample: Transfer 20 µl of glutathione sepharose 4B (Pharmacia) into a 1 ml Eppendorf tube and wash once with 1 ml of PBS, 1% (v/v) Triton X-100, 1 mM EDTA. Add 100 µl of the E. coli lysate to the beads and incubate for 1 hr at room temperature while rotating. After 1 hr wash beads twice with 1 ml of PBS, 1% (v/v) Triton X-100, 1 mM EDTA and remove all supernatant.
B) Pull-down assay
- Transfect cells with 2.5 µg Ras or Rap DNA and 1 µg of Gap protein DNA, using 10 µl Fugene (Roche) in 600 µl serum free medium per φ100 mm dish. Incubate overnight.
- Wash cells twice with PBS (Optional: serum starve cells for 2 hrs in prewarmed imaging buffer at 37 °C). Add 1 ml of ice cold lysis buffer to each dish, detach cells using a cell scraper and transfer lysate to a 1 ml Eppendorf tube. Incubate on ice for 10 min.
- Centrifuge lysate in a microfuge at 4 °C and 15000 rpm for 10 min and discard pellet. Transfer 90 µl of the supernatant to a fresh tube containing 30 µl of 4× SDS PAGE loading dye; add the remainder of the lysate to tube containing 20 µl of the washed GST-RBD beads from the washed GST-RBD beads prepared in section A. Incubate for 1 hr at 4 °C while rotating.
- Pellet beads by centrifugation for 1 min at 6500 rpm in a microfuge and remove supernatant. Add 500 µl of wash buffer (1× PBS, 0.1% (v/v) Triton X-100, 10 mM MgCl2) to each sample and transfer to a 0.5 ml Eppendorf tube. Pellet beads as before and remove supernatant. Wash beads twice more with 500 µl wash buffer and remove all supernatant after final wash.
- Add 15 µl of 4× SDS PAGE loading dye to each sample and adjust volume to a total of 60 µl with wash buffer. (Note: Do not pipette beads to adjust volume, but compare volume to standard tube containing 60 µl liquid.) Boil samples for 5 min and load onto SDS PAGE gels.
Imaging buffer (100 ml)
| Component | Amount |
|---|---|
| NaCl (121 mM) | 3.025 ml of 4 M stock |
| KCl (5.4 mM) | 540 µl of 1 M stock |
| MgCl2 (1.6 mM) | 160 µl of 1 M stock |
| NaHCO3 (6 mM) | 0.0504 g |
| glucose (9 mM) | 405 µl of 40% (w/v) stock |
| Hepes (25 mM) | 2.5 ml of 1 M stock |
| CaCl2 (13 mM) | 130 µl of 1 M stock |
pH to 7.4 with NaOH.
Lysis buffer
- 250 µl of 4 M NaCl
- 500 µl of 1 M Hepes
- 20 µl of 0.5 M EGTA, pH 8.0
- 100 µl of 1 M MgCl2
- pH to 7.5 with NaOH and add H2O to 9.75 ml
Add fresh:
- 0.1 ml Triton X-100
- 0.1 ml of 0.1 M DTT
- 5 µl each of 10 mg/ml benzamidine, trypsin inhibitor, leupeptin, aprotinin, pepstatin A and antipain
- 20 µl of 0.25 M PMSF
Ras binding assay (Kelley et al., EMBO J. 2001)
Alternate pull-down after Kelley, G.G. et al. (EMBO J. 2001): GTP loading in RIPA buffer followed by incubation with GST fusion protein on beads.
- For GTP loading: lyse cells in 700 µl RIPA buffer, spin, use 500 µl, add 10 µl 0.5 M EDTA, 5 µl GDP (or Sigma GDP … or GTPγS, Upstate), incubate at 30 °C for 30 mins. Stop reaction on ice and add 30 µl 1 M MgCl2; continue pull-down.
- Resuspend freshly purified GST fusion proteins to a 50% slurry using RIPA buffer. Incubate a 20 µl portion of this slurry (typically 500 pmol of fusion protein) 30 mins with 500 µl Ras cell lysate overexpressing Ras at 4 °C; retain ~100 µl cell lysate for analysis.
- Following the incubation, centrifuge the reaction at 500 g for 7 min and then wash three times with 600 µl of RIPA buffer. Add a 40 µl aliquot of sample buffer and load 10 µl onto gel. Load ~20 µl cell lysate also to determine relative levels of expression.
RIPA buffer (for 20 ml)
- 10% glycerol — 2 g
- 1% NP-40 — 200 mg
- 50 mM Tris-HCl pH 7.5 — 1 ml 1 M soln. (1 M = 7.85 g/50 ml)
- 200 mM NaCl — 0.8 ml 5 M soln.
- 2 mM MgCl2 — 0.4 ml 1 M soln. (1 M = 10.2 g/50 ml)
- 1 mM PMSF — 200 µl 100 mM soln.
- 5 µg/ml aprotinin
- 5 µg/ml leupeptin
- 10 µg/ml soybean trypsin inhibitor (SBTI). In the absence of SBTI, use benzamidine.