Top 10 tips for successful EMSAs
The electrophoretic mobility shift assay (EMSA), also known as a gel shift assay, remains one of the most widely used methods for studying protein–DNA and protein–RNA interactions. Despite its apparent simplicity, obtaining clean publication-quality data is often tricky and requires careful optimization of probe design, protein quality, binding conditions, and electrophoresis parameters. Many common problems, including weak shifts, smeared bands, high background, and poor reproducibility, can usually be traced back to a handful of experimental variables.
The following ten tips are ones that we have found helpful in producing good quality EMSA data at the Bond Lab. Hopefully these will help you maximize the likelihood of obtaining robust and publication-quality EMSA results.
-
1. Start with High-Quality Protein
The quality of the protein preparation is arguably the single most important determinant of EMSA success. Proteins that are partially denatured, contain detergent or high salt concentrations, are aggregated, or degraded frequently fail to bind their target nucleic acid efficiently. Generally, two types of protein sample are used. These are mammalian cell culture nuclear extracts and purified recombinant proteins. Using nuclear extracts is generally more challenging due to the difficulty in extracting nuclear protein without damaging them. Usually this involves a high salt extraction step but this means that the sample then needs dialysis or dilution or desalting to reduce the salt concentration. Over expression of the factor of interest can often help with detecting binding but this doesn’t allow the natural physiological regulation of that factor to be studied. Purified recombinant proteins can be easier to use as they can be made at much higher concentrations but of course, they do not allow the physiological regulation to be studied.
Whenever possible:
- Use freshly purified protein or nuclear extract
- Avoid repeated freeze-thaw cycles.
- Include reducing agents if required for protein stability.
- Use minimal or no detergent if possible
- Try to prepare your sample in physiological salt levels
If a shift suddenly disappears after previously successful experiments, protein activity should be one of the first factors investigated.
-
2. Design Probes Carefully
Probe design can strongly influence both binding specificity and band interpretation. Ideally, probes should contain a single well-defined binding site and avoid sequences capable of forming extensive secondary structures.
For DNA EMSAs:
- Use short, well-characterized binding regions.
- Minimize additional transcription factor motifs.
- Prepare mutant probes for specificity controls.
- We typically use 5′ biotinylated probes with a TEG spacer
-
3. Optimize the Protein-to-Probe Ratio
One of the most common mistakes is using either too much or too little protein. Excess protein often produces smearing, aggregation, or complexes trapped in the wells, whereas insufficient protein yields weak or undetectable shifts.
Perform a protein titration series spanning at least one order of magnitude. A successful EMSA should show a progressive increase in shifted complex formation as protein concentration increases.
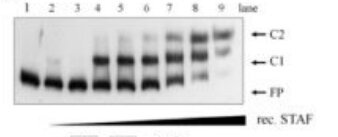
Optimization of purified recombinant STAF/ZNF143 concentrations -
4. Include Non-Specific Competitors
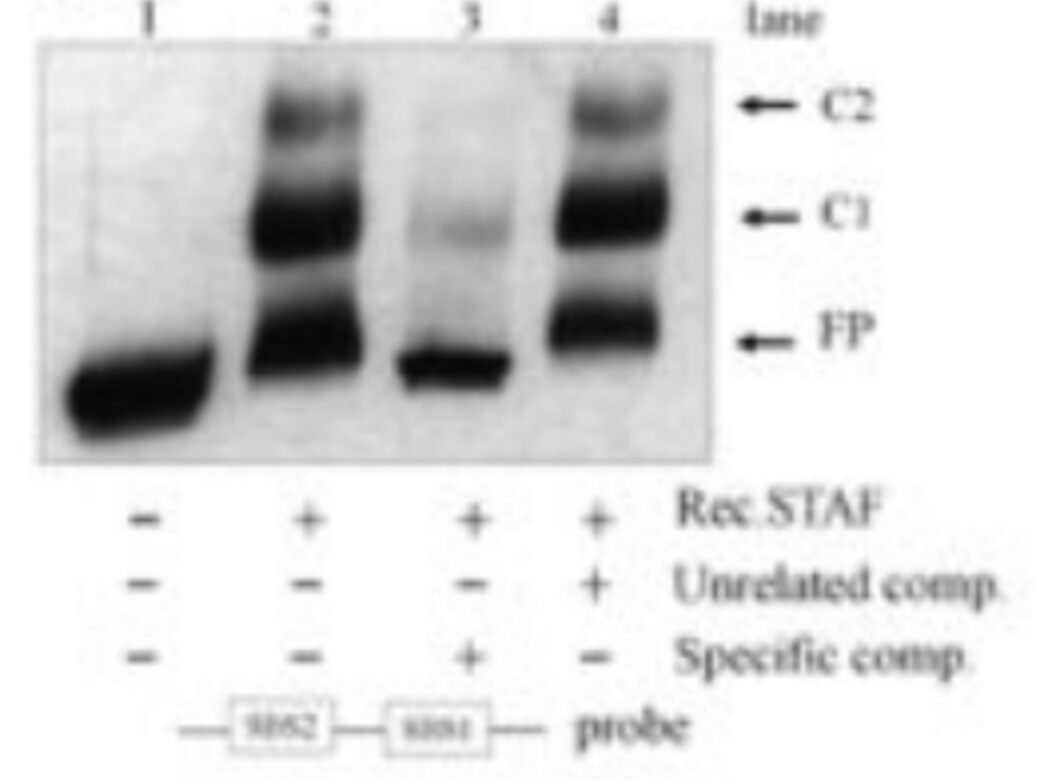
Many DNA-binding proteins exhibit some degree of non-sequence specific nucleic acid affinity. Including competitor nucleic acids such as poly(dI-dC), heparin, or salmon sperm DNA can substantially reduce background binding.
Competitors should generally be added before the labelled probe to occupy non-specific binding sites and improve discrimination of sequence-specific interactions. Also, include controls that include a molar excess (e.g. 20 fold excess) of unlabelled probe that matches the sequence of the labelled probe (should compete with the labelled probe for binding) or a scrambled sequence (that should not compete).
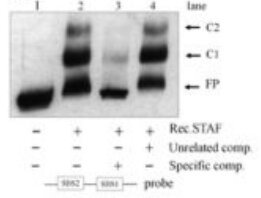
STAF/ZNF143 EMSA with unlabelled probe competitions -
5. Pay Attention to Buffer Composition
Binding buffers frequently require optimization for each protein.
Critical variables include:
- Salt concentration
- pH
- Magnesium concentration
- Reducing agents
- Glycerol content
- Detergents
Low salt conditions can increase non-specific interactions, whereas excessive salt can abolish specific binding. Small changes in ionic strength often have dramatic effects on EMSA performance. Too much glycerol in the sample or loading buffer can cause streaking or smearing of the probe up the gel. We often use Ficoll instead of glycerol in loading buffer because of this.
-
6. Allow Binding Equilibrium to Be Reached
Insufficient incubation time may prevent complexes from reaching equilibrium before electrophoresis.
As a starting point:
- Incubate binding reactions for 20–30 minutes.
- Perform reactions at a temperature appropriate for the biological interaction.
- Consider longer incubations for weakly interacting systems.
For quantitative EMSA experiments, equilibrium binding conditions are particularly important.
-
7. Run Native Gels Under Conditions That Preserve Complexes
Unlike SDS-PAGE, EMSA relies on maintaining intact complexes during electrophoresis.
Important considerations include:
- Use native polyacrylamide gels (absolutely no SDS)
- Avoid overheating during electrophoresis (run in cold room or with cooling)
- Use pre-chilled running buffers when necessary.
- Run gels in a cold room for unstable complexes.
- Allow gels to fully polymerize before use as residual unpolymerized acrylamide can disrupt complex formation. We cast the gels the day before use.
- Pre run gels for about 30 mins before loading samples.
Complex dissociation during electrophoresis often appears as diffuse or smeared bands.
-
8. Choose the Correct Gel Percentage
Gel percentage influences both resolution and complex stability.
General guidelines:
Recommended native gel percentages for EMSA complex size Complex Type Recommended Gel Small oligonucleotides 6–10% PAGE Medium-sized complexes 4–6% PAGE Very large complexes 3–4% PAGE (can be very soft and hard to handle) or native agarose If complexes fail to enter the gel or remain near the wells, reducing gel percentage may improve migration.
-
9. Always Include Proper Controls
Controls transform an observed shift into convincing evidence of specific binding.
Essential controls include:
- Free probe only
- Probe plus protein
- Excess unlabelled specific competitor
- Non-specific competitor
- Mutant probe
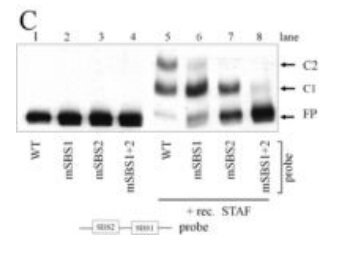
STAF EMSA with mutated probe competition Where antibodies are available, supershift experiments can provide additional confirmation of complex identity.
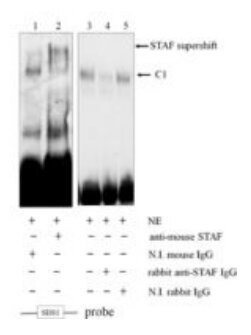
STAF/ZNF143 supershift EMSA -
10. Document Everything During Optimization
EMSA conditions are often highly system dependent. Small changes in buffer composition, salt concentration, probe preparation, or electrophoresis conditions can dramatically alter results.
Maintain detailed records of:
- Protein batch
- Probe preparation and concentration used
- Binding buffer composition (particularly pH, salts and glycerol levels)
- Competitor concentrations
- Gel percentage
- Running conditions (Too high a voltage can cause heating)
- Imaging and exposure settings
Careful documentation greatly accelerates troubleshooting and improves reproducibility between experiments and researchers.
Conclusion
Successful EMSA experiments depend on preserving stable protein–nucleic acid complexes while minimizing non-specific interactions and experimental artifacts. In practice, the greatest improvements usually come from optimizing protein quality, probe design, competitor usage, buffer composition, and electrophoresis conditions. By systematically addressing these variables and incorporating rigorous controls, researchers can obtain clear, reproducible mobility shifts that provide strong evidence for biologically relevant binding interactions.